Methods for mtDNA tracing
Our lab has been active in the development of new technologies to enable lineage tracing in humans via mitochondrial DNA mutations. Most of these works have been collaborative efforts with the Leif Ludwig Lab while we were trainees with Aviv Regev and Vijay Sankaran. See a summary of these major technologies, software, biological results, and informal guidelines below.
Interested in mitochondrial tracing?
A good review on these principles, technologies, and applications is available online as a review article. Feel free to reach out to Caleb, who can either answer your questions or refer you to the right person who can help!
At a high level, we can detect somatic mitochondrial mutations in either mitochondrial DNA (mtDNA) or mtRNA. These methods that we’ve been involved in developing are summarized here:
Mitochondrial single-cell ATAC-seq
The mitochondrial single-cell ATAC-seq (mtscATAC-seq) protocol is an adaptation of commercial single-cell ATAC-seq workflows. At a basic level, by fixing and permeabilizing cells (versus just nuclei), Tn5 also tagments the more accessible mitochondrial genome. Accessible chromatin profiling can thereby be combined with single-cell whole mitochondrial genome sequencing.

ASAP-seq and DOGMA-seq
Enter the ATAC with select antigen profiling by sequencing (ASAP-seq), developed with Eleni Mimitou, Kelvin Chen, and Peter Smibert. ASAP-seq enables a multimodal readout that simultaneously profiles accessible chromatin, protein levels, and mitochondrial DNA mutation for cellular clonality by repurposing existing antibody-oligonucleotide conjugates.
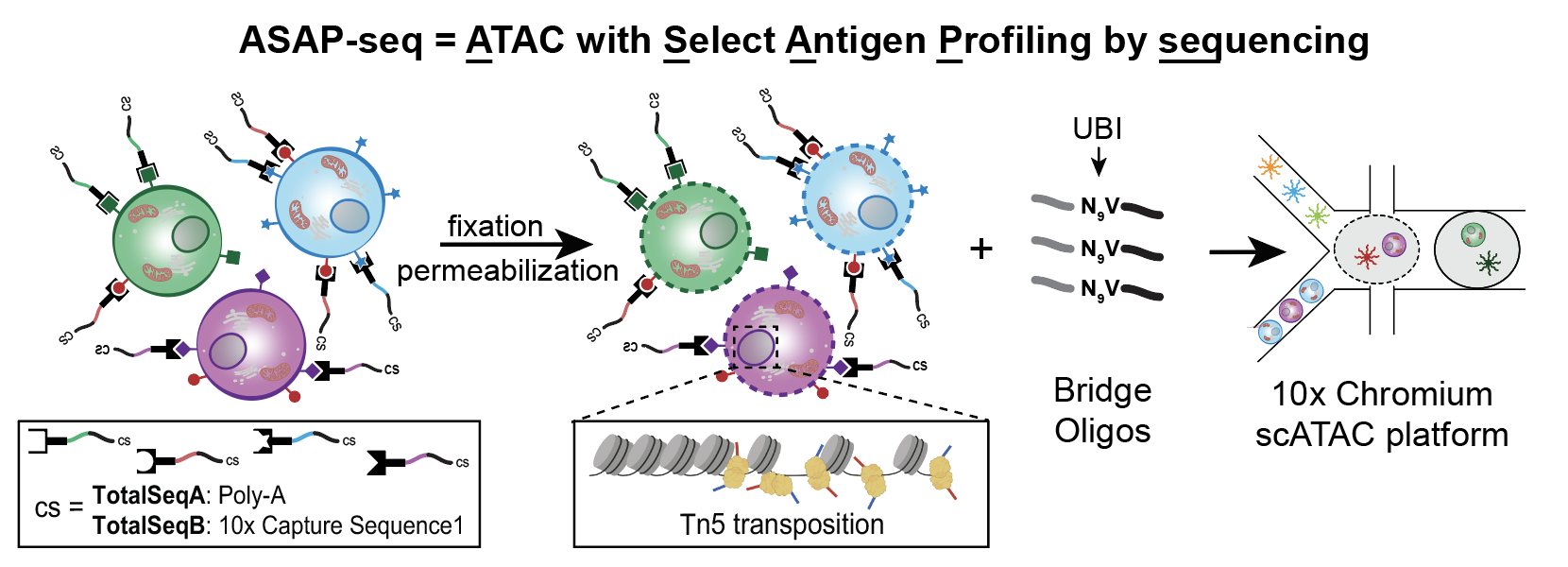
The key insight that unlocked ASAP-seq is the use of a ‘bridge oligo’ for barcoding CITE-seq reagents with gel beads. Specifically, the bead oligo capture sequences on the scATAC-seq kit are complementary to the Nextra Read 1 sequence attached to DNA fragments after Tn5 transposition. Thus, the poly-A CITE-seq reagents are not immediately compatible. The ASAP-seq bridge oligo is complementary to the poly-A antibody-oligo sequence on one end and the Read 1 sequence on the other. This development allows existing CITE-seq reagents to be used directly in ASAP-seq without requiring a new set of antibodies to be conjugated for capture with the ATAC-seq kit.
The 10x Genomics Multiome kit has further facilitated the integration of transcriptional profiles, yielding DOGMA-seq; this enables the single-cell capture of up to four data modalities, including transcriptome, accessible chromatin, surface markers and mtDNA mutations.
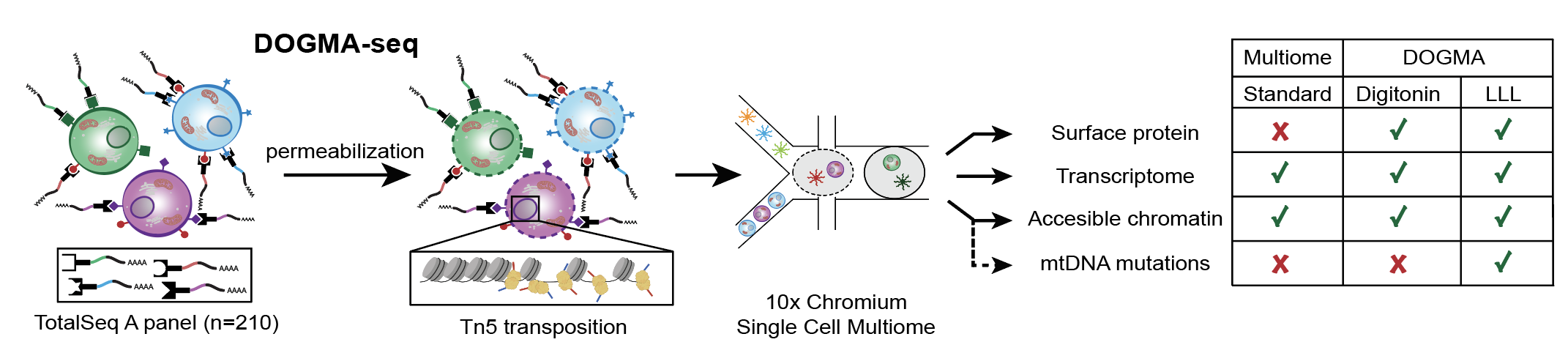
Here, the detection of the oligo-conjugated antibodies is identical to CITE-seq as there are polyT capture sequences on the multiome beads.
PHAGE-ATAC
PHAGE-ATAC (with Evgenij Fiskin), which in addition facilitate surface-marker profiling. In our experience, these approaches both work well and right-out-of-the-box if you have experience working with antibodies and/or phages.
PHAGE-ATAC utilizes an M13 bacteriophage system that genetically encoded antibody binders attached to a PAC-tag, obviating the need for recombinantly expressed antibodies or covalent conjugations. The M13 phagemid system uses nanobodies with known specificity to detect surface antigens and quantify binding in droplets via the barcoding of the CDR3 hypervariable region that directly encodes the protein binder.
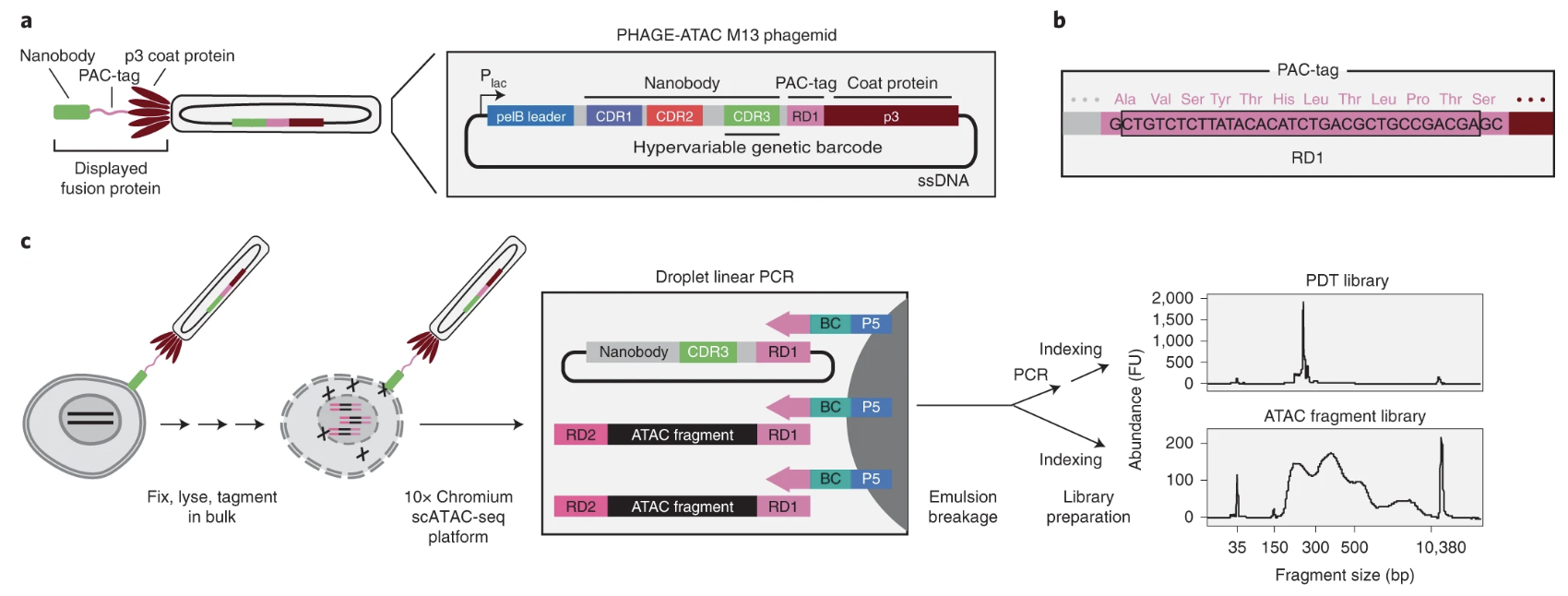
As phages are effectively a renewable resource after infection in bacteria, these reagents are ideal for large-scale experiments. Keep an eye on this assay as additional resources descrbing nanobodies against more targets are developed, including in viral infections like SARS-CoV-2 as we show in the PHAGE-ATAC paper.
Unless you have a specific reason to want the transcriptome (then use DOGMA-seq or MAESTER, below) or the surface proteome (then use ASAP-seq due to ease of commercially available reagents), we recommend mtscATAC-seq due to the ease and cost-efficiency of the workflow.
Another extension of the mtscATAC-seq assay (though unrelated to our group) for the genotyping of targeted loci with single-cell chromatin accessibility (GoT-ChA) utilizes targeted primers to genotype nuclear variants in addition to optional mtDNA sequencing for high-resolution clonal analyses.
MAESTER
As >90% of mtDNA is transcribed, full-length RNA-seq techniques (e.g., Smart-seq) that capture the entire sequence of a transcript are particularly attractive to capture genetic variants. As relatively few groups use Smart-seq nowadays, approaches that are compatible with the more high-throughput and more popular 3′- or 5′-based scRNA-seq approaches were necessary. However, it’s important to note that these approaches sequence only part of the transcript and thus tend to suffer from limited coverage of the mitochondrial transcriptome for confident variant detection.
Enter (MAESTER). MAESTER can enrich mitochondrial mutations from existing cDNA libraries, massively improving the suitability of these data for detecting and analyzing mtDNA variants from high-throughput scRNA-seq data. MAESTER was the brain child of Tyler Miller and Peter van Galen, and we were happy to help out in finalizing its development.

In general, if you are regular user of 10x scRNA-seq, MAESTER may be a great entry-level assay for looking at mitochondrial genomes in your cells However, there are a few noteworthy limitations of genotyping mitochondrial mutations from RNA alone. First, note a high degree of inherent transcriptional error of the mitochondrial RNA polymerase, which we have observed will lead to false-positive heteroplasmy detection. Additionally, not all mtDNA molecules may be equally transcribed or captured, including the 22 mitochondrial tRNAs are not reverse transcribed by poly(A)-based priming. We advise to download public data related to the assay that you plan to use and compute the per-singe-cell coverage to get an estimate of what your data may look like.
One of the best applications of MAESTER resolved the malignant evolution in Barrett’s esophagus (from Syd Shaffer’s Lab) providing a unified view of the cell states and dynamics along a disease trajectory.
Purifying selection in congenital mitochondrial disease
While most single-cell multi-omic efforts so far have used mtDNA genotyping for purposes of clonal tracing, the obvious utility of these technologies for studies of mitochondrial genetics has only become evident more recently in the context of mitochondriopathies. Clinically, these heterogeneous diseases are well recognized, but a detailed molecular understanding of how alterations in mitochondrial genetic integrity elicit changes in individual cells.
We initially applied mtscATAC-seq-based analysis of peripheral blood mononuclear cells from adult patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), who carry the pathogenic m.3243G>A mtDNA variant. In collaboration with Vamsi Mootha and Melissa Walker, We found an almost complete absence of the mutation, specifically in T cells but not in other immune cells.
With Suneet Agarwal, we further applied mtscATAC-seq, ASAP-seq, and DOGMA-seq to study Pearson Syndrome.
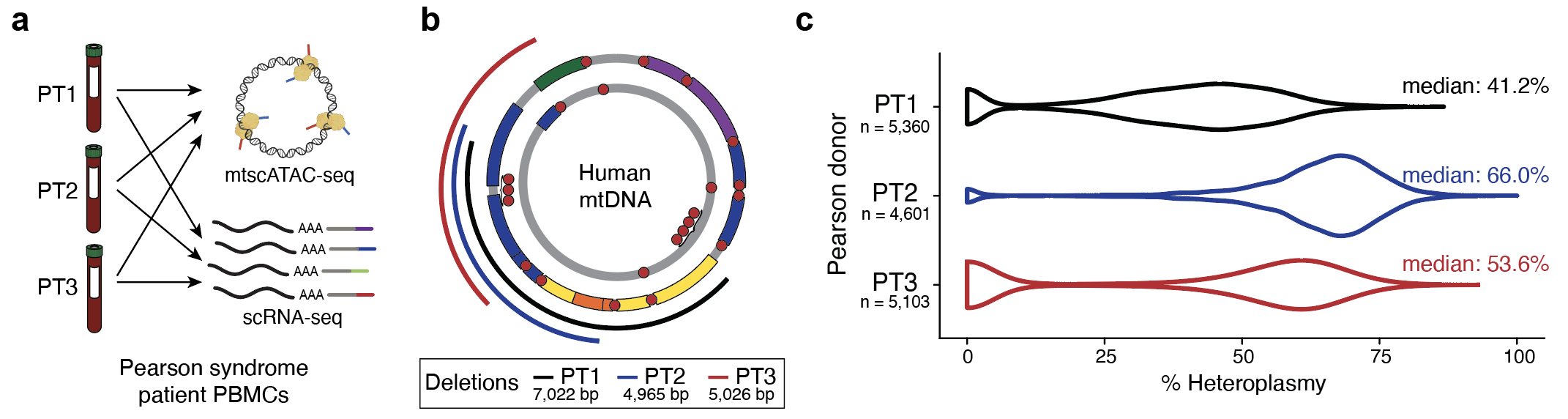
This work showed a strong purifying selection of pathogenic mtDNA in cell states that probably reflect specific metabolic requirements, in particular toward attaining and/or also potentially maintaining the mucosal-associated invariant T cell (MAIT) and CD8+ effector/memory T cell states.
In short, if you want to learn more about how functional mtDNA variants impact cells, the above suite of technologies has worked well in our hands!
Software that enables mitochondrial mutaiton analyses
As part of these tools, our lab has been the primary developers of genotyping software for processing sequencing reads from the protocols and identifying high-confidence single-cell heteroplasmy.
- For analyzing mtDNA (from any ATAC data), we recommend the mgatk package.
- To call mtDNA deletions and quantify single-cell heteroplasmy, we recommend the mgatk-del utility part of mgatk. A detailed summary of the toolkit is here
- For analyzing MAESTER/mRNA data, we recommend the maegatk workflow.
The major differences between the software tools are described here.